
Thalassemia, often abbreviated as THAL, is a group of inherited blood disorders that affect the production of hemoglobin, the protein in red blood cells responsible for carrying oxygen throughout the body. This condition can lead to anemia, fatigue, and other serious health complications. In this article, we will explore the different types of thalassemia, its causes, symptoms, and available treatments to provide a comprehensive understanding of this disorder.
What is Thalassemia?
Thalassemia is a genetic blood disorder characterized by abnormal hemoglobin production. Hemoglobin is made up of two proteins: alpha globin and beta globin. When there is a defect in the genes responsible for producing these proteins, it results in reduced or absent hemoglobin, leading to fewer healthy red blood cells. This can cause mild to severe anemia depending on the type and severity of the condition.
How Common is Thalassemia?
- Thalassemia is most commonly found in people of Mediterranean, Middle Eastern, African, South Asian, and Southeast Asian descent.
- It is estimated that millions of people worldwide are carriers of thalassemia, with thousands of children born with the condition each year.
Types of Thalassemia
There are several types of thalassemia, which are classified based on the specific part of the hemoglobin molecule affected and the severity of the condition.
Alpha Thalassemia
Alpha thalassemia occurs when there is a defect in the genes responsible for producing alpha globin. The severity of the condition depends on how many of the four alpha globin genes are affected:
- Silent Carrier: Only one gene is affected, and the person usually shows no symptoms.
- Alpha Thalassemia Trait: Two genes are affected, leading to mild anemia.
- Hemoglobin H Disease: Three genes are affected, resulting in moderate to severe anemia.
- Hydrops Fetalis: All four genes are affected, causing life-threatening anemia before birth.
Beta Thalassemia
Beta thalassemia occurs when there is a defect in the genes responsible for producing beta globin. The severity of the condition depends on whether one or both genes are affected:
- Beta Thalassemia Minor: Only one gene is affected, leading to mild anemia.
- Beta Thalassemia Intermedia: Both genes are affected, but the condition is less severe than major thalassemia.
- Beta Thalassemia Major: Both genes are affected, causing severe anemia that requires regular blood transfusions.
Causes of Thalassemia
Thalassemia is caused by mutations in the genes responsible for hemoglobin production. These mutations are inherited from one or both parents. Understanding the inheritance patterns can help determine the risk of passing the condition to future generations.
Inheritance Patterns
- Autosomal Recessive Inheritance: Thalassemia is inherited in an autosomal recessive pattern, meaning both parents must pass on a defective gene for their child to develop the condition.
- Carrier Status: If only one parent passes on the defective gene, the child becomes a carrier and may have mild symptoms or none at all.
Risk Factors
- Family history of thalassemia increases the risk of inheriting the condition.
- Certain ethnic groups are more likely to carry the defective genes due to higher prevalence in their populations.
Symptoms of Thalassemia
The symptoms of thalassemia vary depending on the type and severity of the condition. Some individuals may experience mild symptoms, while others may face life-threatening complications.
Mild Symptoms
- Fatigue and weakness due to mild anemia.
- Pale or yellowish skin.
- Mild enlargement of the spleen.
Severe Symptoms
- Severe anemia requiring frequent blood transfusions.
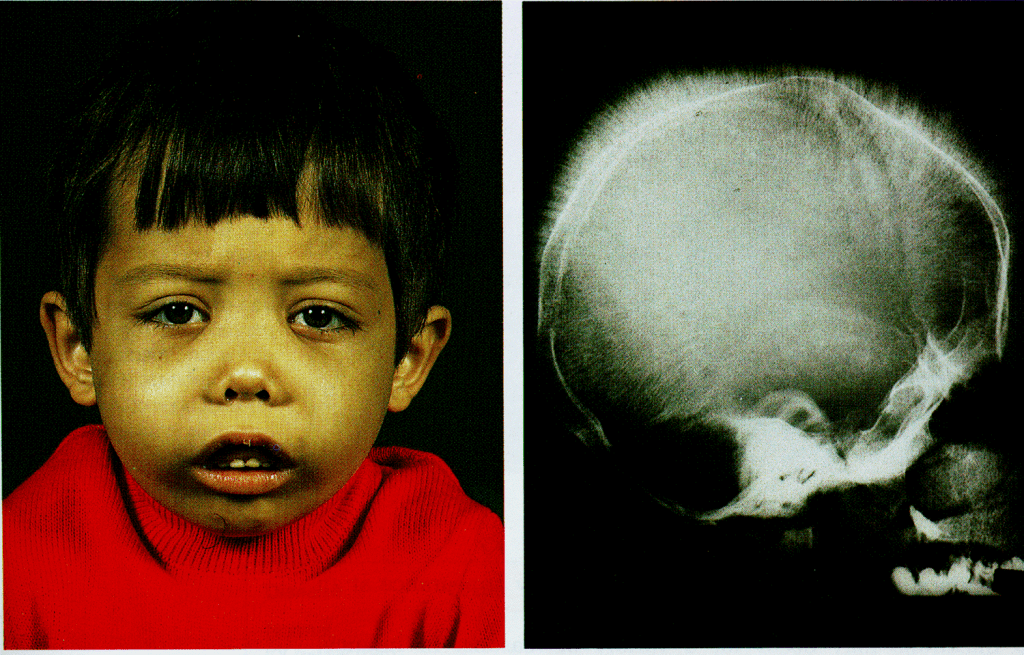
- Bone deformities, especially in the face, due to bone marrow expansion.
- Growth delays in children.
- Dark urine caused by the breakdown of red blood cells.
Treatments for Thalassemia
While there is no cure for thalassemia, various treatments are available to manage the symptoms and improve the quality of life for those affected. The treatment plan depends on the type and severity of the condition.
Blood Transfusions
For individuals with severe thalassemia, regular blood transfusions are necessary to maintain healthy levels of red blood cells. Blood transfusions help alleviate symptoms of anemia and prevent complications.
Iron Chelation Therapy
Frequent blood transfusions can lead to iron overload in the body, which can damage organs such as the heart and liver. Iron chelation therapy involves medications that remove excess iron from the body.
Bone Marrow Transplant
A bone marrow transplant, also known as a stem cell transplant, is the only potential cure for thalassemia. This procedure replaces defective bone marrow with healthy donor marrow, allowing the body to produce normal hemoglobin.
Medications and Supplements
Supplements such as folic acid can support red blood cell production. Additionally, medications may be prescribed to manage complications like infections or bone pain.
Lifestyle Modifications
- Avoiding iron-rich foods and supplements to prevent iron overload.
- Regular monitoring of iron levels and organ function through blood tests.
- Adopting a healthy diet and staying physically active to support overall well-being.
Living with Thalassemia
Managing thalassemia requires a multidisciplinary approach involving healthcare providers, family support, and lifestyle adjustments. Early diagnosis and consistent medical care play a crucial role in improving outcomes for individuals with thalassemia.
Emotional and Psychological Support
- Counseling and support groups can help individuals and families cope with the emotional challenges of living with thalassemia.
- Educating oneself about the condition empowers patients to make informed decisions about their care.
Preventive Measures
- Genetic counseling is recommended for couples planning to have children, especially if they are carriers of thalassemia.
- Prenatal testing can identify thalassemia in unborn babies, allowing parents to prepare for the child’s needs.