
Metachromatic leukodystrophy, often abbreviated as MLD, is a rare and progressive genetic disorder that affects the nervous system. This condition belongs to a group of diseases known as lysosomal storage disorders, which are caused by the malfunction of specific enzymes within the body’s cells. In this article, we will delve into the details of this complex disease, exploring its causes, symptoms, diagnosis, treatment options, and ongoing research efforts.
What Is Metachromatic Leukodystrophy?
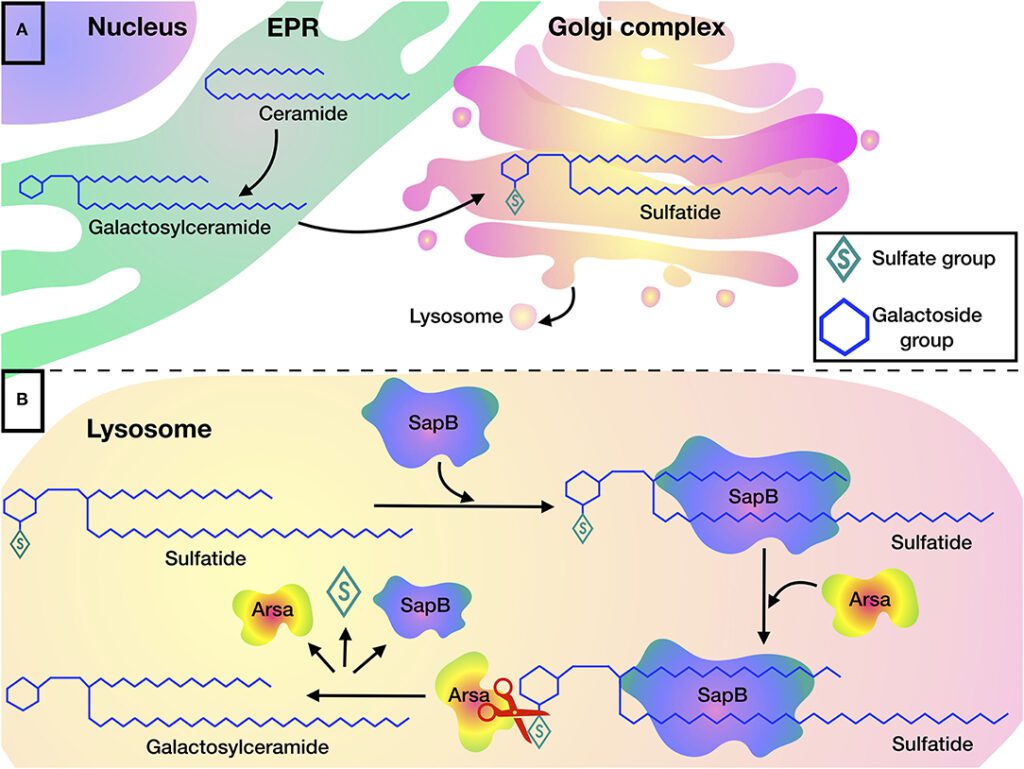
Metachromatic leukodystrophy is a genetic disorder that primarily impacts the central and peripheral nervous systems. It occurs due to the accumulation of sulfatides, a type of fat molecule, in the brain, spinal cord, and peripheral nerves. Over time, this buildup damages the protective covering of nerve fibers, called myelin, leading to the degeneration of these vital structures. As a result, individuals with this condition experience a range of neurological symptoms that worsen over time.
Understanding the Role of Myelin
Myelin is a crucial component of the nervous system, acting as an insulating layer around nerve fibers. It ensures the efficient transmission of electrical signals between neurons, enabling proper communication within the body. When myelin is damaged or destroyed, as it is in metachromatic leukodystrophy, the ability of the nervous system to function effectively is severely compromised.
Causes of Metachromatic Leukodystrophy
This condition is caused by mutations in the ARSA gene, which provides instructions for producing an enzyme called arylsulfatase A. This enzyme is responsible for breaking down sulfatides. When the ARSA gene is mutated, the enzyme’s activity is reduced or absent, leading to the toxic buildup of sulfatides in the body.
Inheritance Pattern
Metachromatic leukodystrophy follows an autosomal recessive inheritance pattern. This means that an individual must inherit two copies of the mutated gene—one from each parent—to develop the disease. Parents who carry one copy of the mutated gene are typically unaffected but have a 25% chance of passing the condition to their child with each pregnancy.
Symptoms of Metachromatic Leukodystrophy
The symptoms of this condition vary depending on the age of onset, which can be categorized into three main forms: late infantile, juvenile, and adult. Each form has distinct characteristics and progression patterns.
Late Infantile Form
- Onset typically occurs between 6 months and 2 years of age.
- Initial symptoms include difficulty walking, muscle weakness, and loss of motor skills.
- As the disease progresses, children may experience seizures, vision loss, hearing impairment, and cognitive decline.
- Most affected children lose the ability to speak and move independently within a few years of symptom onset.
Juvenile Form
- Onset usually occurs between 3 and 16 years of age.
- Early signs may include behavioral changes, declining school performance, and difficulties with coordination.
- Over time, individuals may develop muscle stiffness, seizures, and problems with speech and swallowing.
- Progression is slower compared to the late infantile form, but significant disability often develops within a decade.
Adult Form
- Onset typically occurs after the age of 16.
- Symptoms may begin with psychiatric issues, such as depression or personality changes, followed by motor difficulties.
- Progression is generally slower than in the other forms, but individuals may eventually experience severe neurological impairment.
Diagnosing Metachromatic Leukodystrophy
Diagnosing this condition can be challenging due to its rarity and the variability of symptoms. However, early diagnosis is critical for managing the disease and planning appropriate interventions.
Initial Evaluation
A healthcare provider will typically begin with a thorough medical history and physical examination. They may look for signs of neurological dysfunction, such as muscle weakness, poor coordination, or developmental delays.
Laboratory Tests
To confirm a diagnosis, several specialized tests may be performed:
- Enzyme Activity Assay: This test measures the activity of arylsulfatase A in blood or skin cells. Reduced enzyme activity is indicative of the condition.
- Genetic Testing: Identifying mutations in the ARSA gene can provide definitive confirmation of the diagnosis.
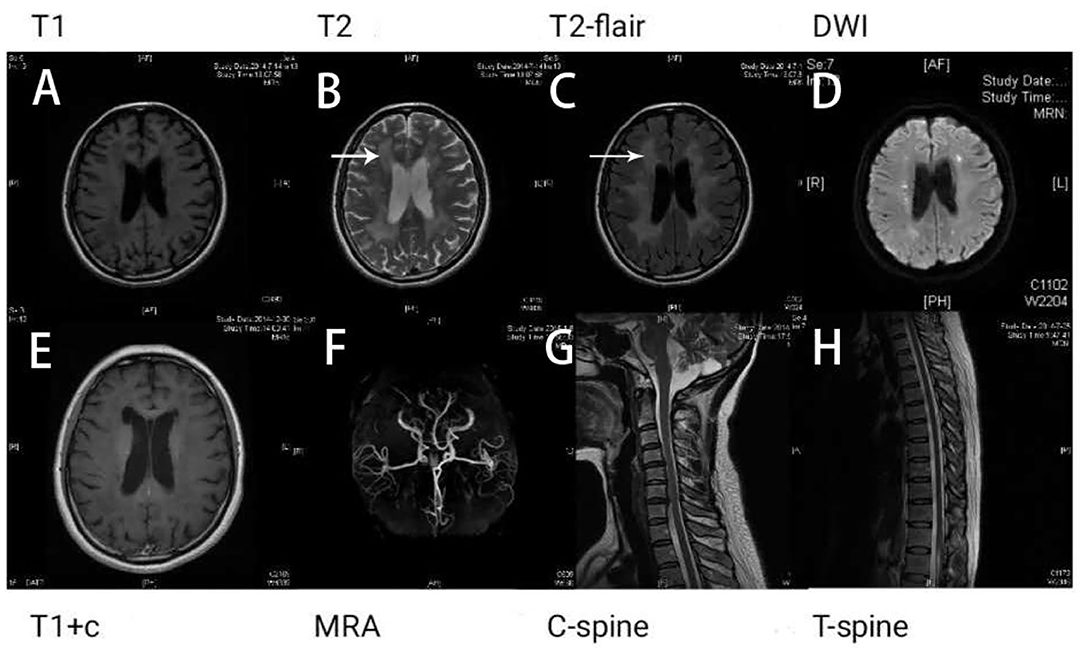
- Imaging Studies: Magnetic resonance imaging (MRI) of the brain may reveal characteristic changes in white matter, which can support the diagnosis.
Treatment Options for Metachromatic Leukodystrophy
Currently, there is no cure for this condition. However, several treatment approaches aim to manage symptoms, slow disease progression, and improve quality of life.
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation, also known as bone marrow transplantation, is the most widely used treatment for this condition. This procedure involves replacing defective cells with healthy donor cells capable of producing the missing enzyme. While it can slow disease progression, it is most effective when performed before significant neurological damage has occurred.
Gene Therapy
Recent advancements in gene therapy offer promising new possibilities for treating this condition. This approach involves introducing a functional copy of the ARSA gene into the patient’s cells, enabling them to produce the missing enzyme. Clinical trials have shown encouraging results, particularly in younger patients with early-stage disease.
Symptomatic and Supportive Care
Managing symptoms is a key component of care for individuals with this condition. This may include:
- Physical therapy to maintain mobility and prevent contractures.
- Medications to control seizures and muscle stiffness.
- Nutritional support to address swallowing difficulties and ensure adequate nutrition.
- Psychological and psychiatric support to address emotional and behavioral challenges.
Ongoing Research and Future Directions
Research into this condition is ongoing, with scientists exploring new ways to understand and treat the disease. Some areas of focus include:
Advancements in Gene Therapy
Gene therapy holds significant promise for addressing the root cause of this condition. Researchers are working to refine techniques and expand access to this treatment option.
Development of Enzyme Replacement Therapy
Efforts are underway to develop enzyme replacement therapies that can deliver arylsulfatase A directly to affected cells. While challenges remain, this approach could provide a less invasive alternative to stem cell transplantation.
Biomarker Discovery
Identifying reliable biomarkers for this condition could improve early diagnosis and enable better monitoring of disease progression and treatment response.
Raising Awareness and Supporting Affected Families
Given the rarity of this condition, raising awareness is essential for ensuring timely diagnosis and access to care. Patient advocacy groups play a crucial role in connecting families, providing resources, and funding research initiatives. By fostering collaboration among researchers, clinicians, and affected communities, progress can be made toward improving outcomes for individuals with this condition.