
Amyloidosis, often abbreviated as AL for amyloid light chain in medical contexts, is a rare and complex group of diseases characterized by the abnormal buildup of proteins in various tissues and organs. This condition disrupts normal organ function and can lead to severe health complications if left untreated. The term “amyloid” refers to the misfolded proteins that aggregate and deposit in organs such as the heart, kidneys, liver, and nervous system. Understanding the underlying causes, recognizing the symptoms, and exploring treatment options are essential steps in managing this challenging condition.
What Is Amyloidosis?
Amyloidosis occurs when the body produces abnormal proteins that cannot be broken down or eliminated properly. These proteins clump together and form insoluble fibrils, which then accumulate in tissues and organs. Over time, these deposits interfere with the normal functioning of the affected organs, leading to a wide range of symptoms and complications.
There are several types of amyloidosis, each classified based on the specific protein involved and the organs affected. Some forms of amyloidosis are systemic, meaning they affect multiple organs, while others are localized, impacting only one area of the body. Regardless of the type, the disease poses significant challenges for both patients and healthcare providers due to its complexity and variability.
Types of Amyloidosis
- Primary Amyloidosis: This form is associated with a plasma cell disorder and involves the deposition of light chain proteins produced by abnormal plasma cells in the bone marrow.
- Secondary Amyloidosis: This type occurs as a complication of chronic inflammatory diseases, such as rheumatoid arthritis or inflammatory bowel disease, and involves the accumulation of a different protein called amyloid A.
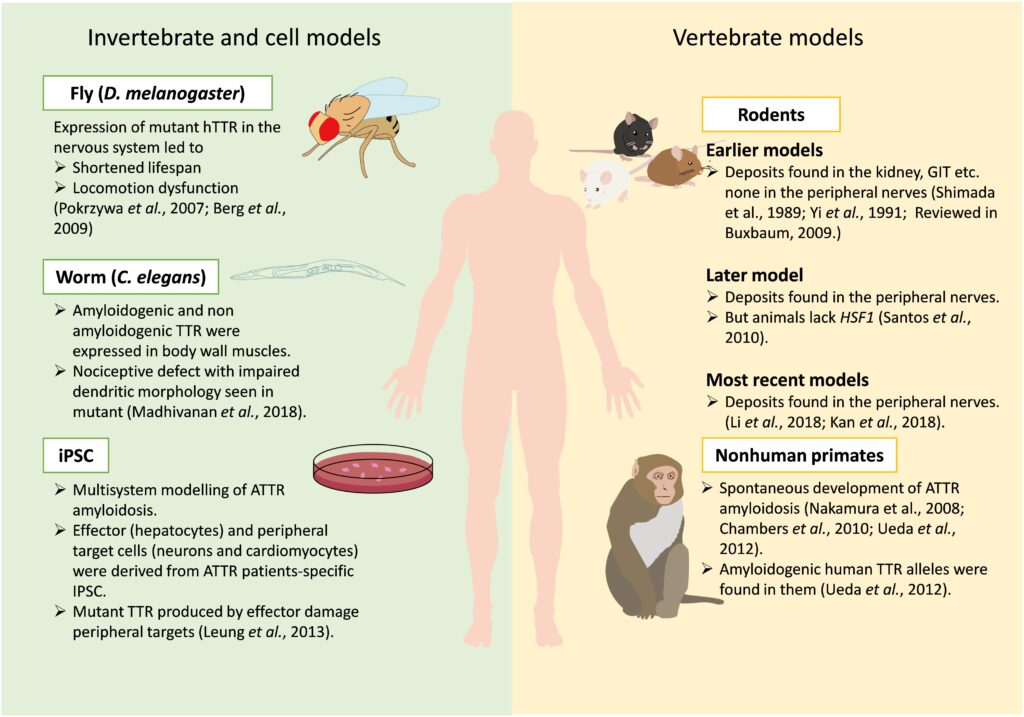
- Familial Amyloidosis: This is an inherited form of the disease caused by genetic mutations that lead to the production of abnormal proteins.
- Dialysis-Related Amyloidosis: This form affects individuals who have been on long-term dialysis and involves the buildup of beta-2 microglobulin protein in joints and tendons.
Causes of Amyloidosis
The exact cause of amyloidosis depends on the type of the disease. In primary amyloidosis, the root cause lies in the bone marrow, where abnormal plasma cells produce excessive amounts of light chain proteins. These proteins are normally part of antibodies, but when produced in excess, they misfold and form amyloid fibrils.
In secondary amyloidosis, chronic inflammation plays a key role. Conditions such as tuberculosis, osteomyelitis, or autoimmune diseases trigger the liver to produce high levels of a protein known as serum amyloid A. Over time, this protein transforms into amyloid deposits in various organs.
Familial amyloidosis is caused by mutations in specific genes that are passed down through families. These mutations result in the production of abnormal proteins that are prone to forming amyloid deposits. The most common mutation affects the transthyretin protein, which is primarily produced in the liver.
Dialysis-related amyloidosis occurs in individuals undergoing long-term hemodialysis. During dialysis, the kidneys’ inability to filter certain proteins leads to their accumulation in the blood. Over time, these proteins deposit in joints and other tissues, causing pain and stiffness.
Risk Factors
- Age: Older adults are at a higher risk of developing amyloidosis.
- Gender: Men are more likely to develop certain types of amyloidosis than women.
- Chronic Diseases: Conditions such as rheumatoid arthritis, tuberculosis, or inflammatory bowel disease increase the risk of secondary amyloidosis.
- Family History: Individuals with a family history of amyloidosis are at a higher risk of developing familial forms of the disease.
- Long-Term Dialysis: Prolonged use of dialysis increases the likelihood of dialysis-related amyloidosis.
Symptoms of Amyloidosis
The symptoms of amyloidosis vary widely depending on the organs affected. Because the disease can impact multiple systems in the body, the presentation of symptoms can be diverse and nonspecific, making diagnosis challenging. Some common symptoms include:
Cardiac Symptoms
- Shortness of breath
- Irregular heartbeat
- Swelling in the legs and ankles
- Fatigue
When amyloid deposits accumulate in the heart, they can cause thickening of the heart walls and impair its ability to pump blood effectively. This can lead to heart failure, arrhythmias, and other serious complications.
Kidney Symptoms
- Swelling in the legs, feet, or around the eyes
- Proteinuria (excess protein in the urine)
- Decreased urine output
- High blood pressure
Kidney involvement is common in amyloidosis and can lead to nephrotic syndrome, a condition characterized by significant protein loss in the urine, swelling, and elevated cholesterol levels.
Liver Symptoms
- Enlarged liver
- Pain or discomfort in the upper right abdomen
- Jaundice (yellowing of the skin and eyes)
When amyloid deposits build up in the liver, it can cause enlargement and impaired liver function, leading to symptoms such as jaundice and abdominal discomfort.
Nervous System Symptoms
- Numbness or tingling in the hands and feet
- Burning pain
- Carpal tunnel syndrome
- Dizziness or fainting upon standing
Amyloid deposits in the nervous system can cause peripheral neuropathy, autonomic dysfunction, and other neurological issues. Autonomic neuropathy, in particular, can lead to problems with blood pressure regulation, digestion, and bladder control.
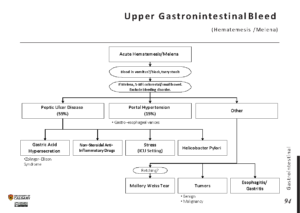
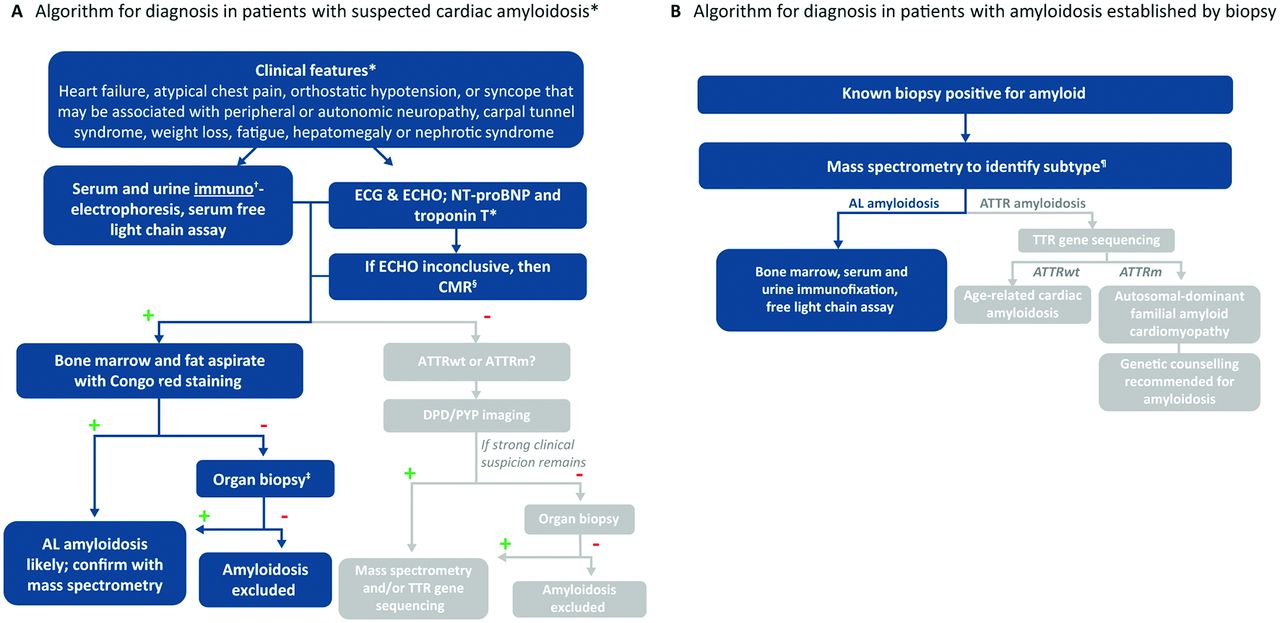
Diagnosis of Amyloidosis
Diagnosing amyloidosis requires a combination of clinical evaluation, laboratory tests, imaging studies, and biopsy. Due to the nonspecific nature of the symptoms, diagnosis can be challenging and often delayed. Key diagnostic steps include:
Biopsy
A biopsy is the gold standard for diagnosing amyloidosis. During this procedure, a small sample of tissue is taken from an affected organ or another accessible site, such as the fat pad under the skin or the rectum. The tissue is then examined under a microscope using special stains to detect the presence of amyloid deposits.
Imaging Studies
Imaging techniques such as echocardiography, magnetic resonance imaging, and computed tomography scans can help assess the extent of organ involvement. For example, an echocardiogram can reveal thickened heart walls and reduced heart function, which are indicative of cardiac amyloidosis.
Laboratory Tests
Blood and urine tests are used to evaluate organ function and detect abnormalities such as proteinuria, elevated liver enzymes, or abnormal levels of specific proteins. Genetic testing may also be performed to identify mutations associated with familial amyloidosis.
Treatment Options for Amyloidosis
While there is no cure for amyloidosis, treatment focuses on managing symptoms, slowing disease progression, and improving quality of life. The specific treatment approach depends on the type of amyloidosis and the organs involved. Common treatment strategies include:
Medications
- Chemotherapy Drugs: Medications such as melphalan and bortezomib are used to target abnormal plasma cells in primary amyloidosis.
- Anti-Inflammatory Drugs: These medications are used to manage inflammation in secondary amyloidosis.
- Heart Medications: Diuretics, beta-blockers, and other drugs may be prescribed to manage heart-related symptoms.
Stem Cell Transplantation
For some patients with primary amyloidosis, high-dose chemotherapy followed by stem cell transplantation may be an option. This procedure aims to eliminate abnormal plasma cells and restore healthy bone marrow function.
Organ Transplantation
In cases where a single organ is severely affected, such as the liver in familial amyloidosis, organ transplantation may be considered. Liver transplantation can halt the production of abnormal transthyretin protein in familial amyloidosis.
Supportive Care
Supportive care plays a crucial role in managing amyloidosis. This includes dietary modifications, physical therapy, pain management, and addressing complications such as kidney failure or heart failure. Regular follow-up with a multidisciplinary team of specialists is essential to monitor disease progression and adjust treatment as needed.
Emerging Therapies and Research
Ongoing research is focused on developing new therapies to target the underlying mechanisms of amyloidosis. Some promising areas of investigation include:
- Gene Therapy: Researchers are exploring ways to correct genetic mutations responsible for familial amyloidosis.
- Monoclonal Antibodies: These targeted therapies aim to bind to and remove amyloid deposits from tissues.
- Small Molecule Drugs: New medications are being developed to stabilize proteins and prevent their misfolding.
Advances in understanding the molecular basis of amyloidosis are paving the way for more effective treatments and improved outcomes for patients.