
Creutzfeldt-Jakob Disease, commonly abbreviated as CJD, is a rare and fatal brain disorder that falls under the category of prion diseases. These diseases are caused by abnormal proteins called prions, which lead to the rapid degeneration of brain tissue. This article provides an in-depth exploration of Creutzfeldt-Jakob Disease, including its causes, symptoms, diagnosis, treatment options, and ongoing research efforts.
Understanding the Basics of Creutzfeldt-Jakob Disease
Creutzfeldt-Jakob Disease is a neurodegenerative condition that affects approximately one in every million people worldwide. It is characterized by the progressive and irreversible destruction of brain cells, leading to severe neurological symptoms. The disease is part of a larger group of disorders known as transmissible spongiform encephalopathies, which also includes conditions like bovine spongiform encephalopathy (commonly referred to as mad cow disease) and scrapie in sheep.
The Role of Prions in the Disease
At the heart of Creutzfeldt-Jakob Disease lies an abnormal protein known as a prion. Unlike bacteria or viruses, prions are not living organisms. Instead, they are misfolded versions of normal proteins found in the brain. When these abnormal proteins come into contact with healthy proteins, they cause them to misfold as well, creating a chain reaction that results in widespread damage to brain tissue. This process leads to the characteristic sponge-like appearance of the brain seen in affected individuals.
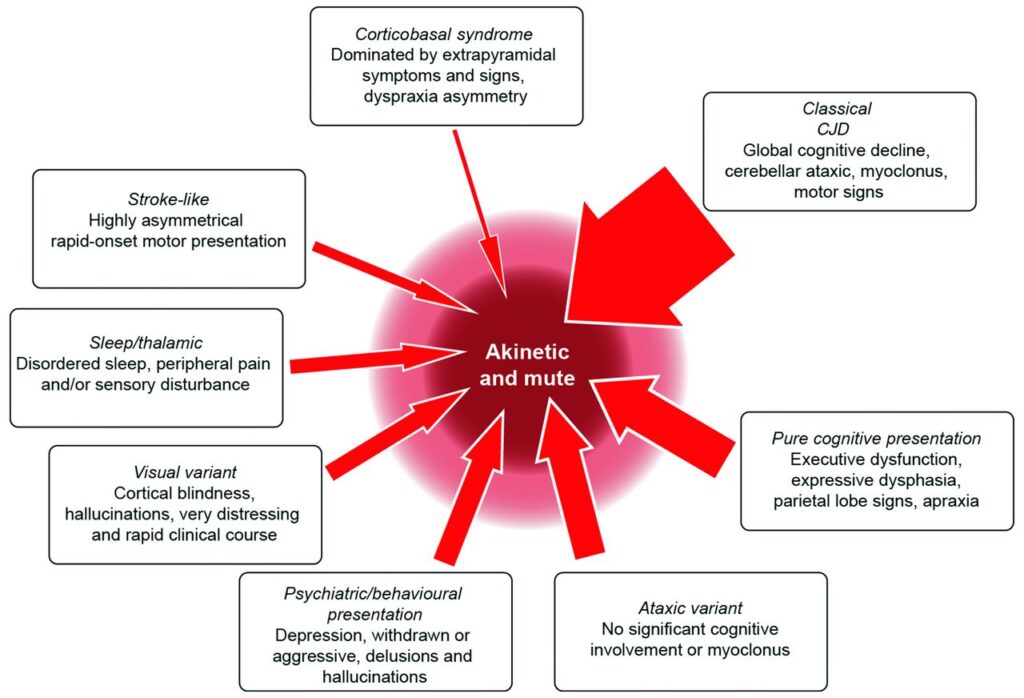
Types of Creutzfeldt-Jakob Disease
There are several forms of this disease, each with distinct characteristics:
- Sporadic Creutzfeldt-Jakob Disease: This is the most common form, accounting for about 85 percent of cases. Its exact cause remains unknown, but it is believed to occur spontaneously due to random mutations in the prion protein gene.
- Familial Creutzfeldt-Jakob Disease: This type is inherited and occurs when there is a mutation in the prion protein gene passed down through families. It accounts for about 10 to 15 percent of cases.
- Acquired Creutzfeldt-Jakob Disease: This form is transmitted through exposure to infected tissue, such as during medical procedures involving contaminated instruments or through consumption of contaminated meat. It is the rarest form of the disease.
Symptoms and Progression of the Disease
The symptoms of Creutzfeldt-Jakob Disease typically appear suddenly and worsen rapidly. Early signs may be subtle and easily mistaken for other neurological conditions, making diagnosis challenging. As the disease progresses, the symptoms become more severe and debilitating.
Early Symptoms
In the early stages, individuals may experience:
- Memory loss and cognitive decline
- Changes in personality and behavior
- Difficulty with coordination and balance
- Blurred vision or blindness
- Insomnia or other sleep disturbances
Advanced Symptoms
As the disease advances, the following symptoms may develop:
- Severe dementia
- Involuntary muscle jerks (myoclonus)
- Loss of the ability to speak or move
- Coma
The progression of the disease is rapid, with most individuals succumbing to it within a year of symptom onset. In some cases, death occurs even sooner, often due to complications such as pneumonia or infections caused by immobility.
Diagnosis of Creutzfeldt-Jakob Disease
Diagnosing this condition is challenging because its symptoms mimic those of other neurological disorders, such as Alzheimer’s disease or Parkinson’s disease. However, certain diagnostic tools and tests can help confirm the presence of the disease.
Clinical Evaluation
A thorough clinical evaluation is the first step in diagnosing this condition. Doctors will review the patient’s medical history, conduct a physical examination, and assess neurological function. They may also inquire about any family history of similar illnesses to determine if the disease could be familial.
Diagnostic Tests
Several tests can aid in confirming the diagnosis:
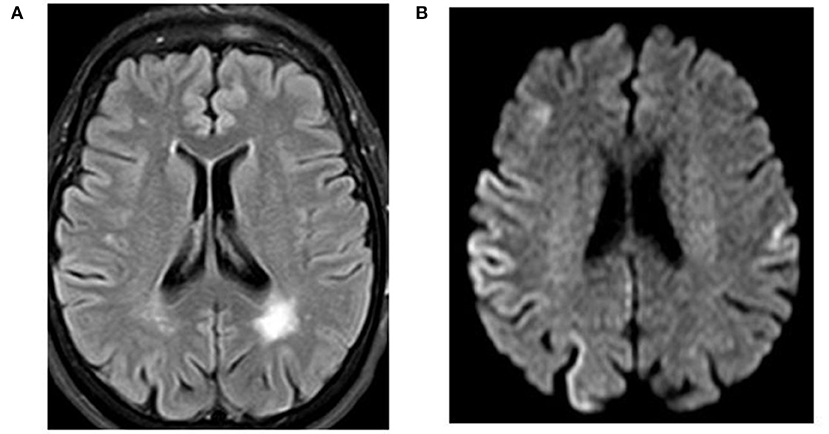
- Magnetic Resonance Imaging (MRI): This imaging technique can reveal characteristic changes in the brain, such as atrophy or signal abnormalities in specific regions.
- Electroencephalogram (EEG): An EEG measures electrical activity in the brain and may show distinctive patterns associated with this disease.
- Cerebrospinal Fluid Analysis: A lumbar puncture can detect elevated levels of certain proteins, such as 14-3-3, which are indicative of rapid neuronal death.
- Brain Biopsy: In rare cases, a biopsy of brain tissue may be performed to confirm the presence of prions. However, this procedure is invasive and carries risks, so it is typically reserved for cases where other tests are inconclusive.
Treatment and Management Options
Currently, there is no cure for this disease. Treatment focuses on managing symptoms and providing supportive care to improve the quality of life for affected individuals and their families.
Medications
While no medications can stop the progression of the disease, certain drugs may help alleviate specific symptoms:
- Anticonvulsants: These medications can help control involuntary muscle jerks and seizures.
- Antidepressants or Antipsychotics: These may be prescribed to manage mood swings, anxiety, or hallucinations.
Supportive Care
Supportive care plays a crucial role in managing the disease. This includes:
- Physical therapy to maintain mobility and prevent complications like bedsores
- Occupational therapy to assist with daily activities
- Nutritional support to address swallowing difficulties and ensure adequate nutrition
- Hospice care to provide comfort and dignity in the final stages of the disease
Ongoing Research and Future Directions
Despite the challenges posed by this disease, researchers are actively working to better understand its mechanisms and develop potential treatments. Several promising areas of research are currently being explored.
Advances in Prion Research
Scientists are investigating the structure and behavior of prions to identify ways to disrupt their harmful effects. Understanding how prions spread and replicate could pave the way for targeted therapies that halt or slow the progression of the disease.
Experimental Treatments
Several experimental treatments are in various stages of development:
- Prion-Binding Molecules: Researchers are testing compounds that can bind to prions and neutralize their toxic effects.
- Gene Therapy: Efforts are underway to use gene-editing technologies to correct mutations in the prion protein gene responsible for familial forms of the disease.
- Vaccines: Although still in the early stages, some studies are exploring the possibility of developing vaccines to prevent prion-related diseases.
Improved Diagnostic Tools
Enhancing diagnostic capabilities is another critical area of focus. New imaging techniques and biomarkers are being developed to enable earlier and more accurate detection of the disease. Early diagnosis could allow for timely interventions and improve outcomes for affected individuals.
Prevention Strategies
While there is no guaranteed way to prevent this disease, certain measures can reduce the risk of acquiring the acquired form:
- Avoid consuming meat from animals suspected of having prion-related diseases.
- Ensure strict sterilization protocols are followed during medical procedures to prevent contamination.
- Screen donated blood and organs for prion-related markers to minimize transmission risks.
For individuals with a family history of the familial form, genetic counseling may provide valuable insights into their risk and guide decisions about family planning.
Raising Awareness and Support
Raising awareness about this disease is essential to promote understanding and support for affected individuals and their families. Advocacy organizations play a vital role in funding research, educating the public, and providing resources for caregivers. By fostering a sense of community, these efforts help reduce the stigma and isolation often associated with rare diseases.