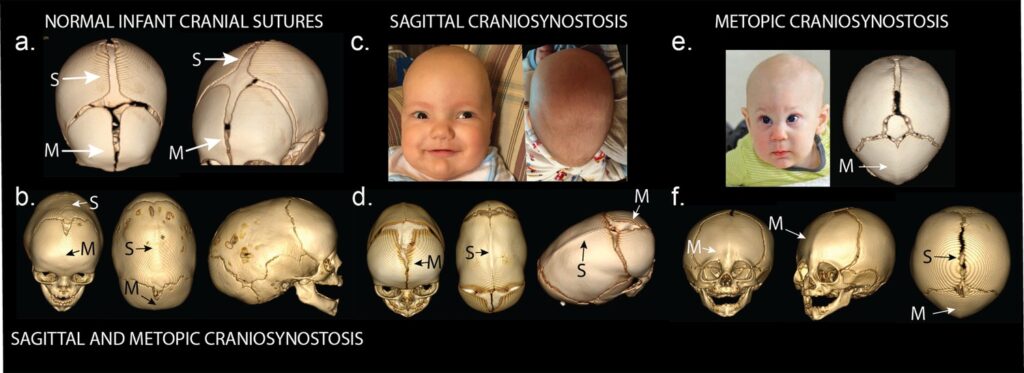

Craniosynostosis, often abbreviated as CS, is a medical condition characterized by the premature fusion of one or more sutures in an infant’s skull. This abnormal fusion disrupts the natural growth and development of the skull, potentially leading to various physical and neurological complications. In this article, we will explore the intricacies of this condition, including its causes, types, symptoms, diagnosis, and treatment options.
Understanding the Basics of Cranial Sutures
The human skull is composed of several bones that are initially separate at birth. These bones are connected by flexible fibrous joints called sutures. The primary purpose of these sutures is to allow the skull to expand and accommodate the rapid brain growth that occurs during infancy. Normally, these sutures remain open and flexible until the child reaches early adulthood, when they eventually fuse. However, in cases of craniosynostosis, one or more sutures close prematurely, restricting the skull’s ability to grow properly.
Types of Craniosynostosis
There are several types of craniosynostosis, each classified based on the specific suture or sutures affected. Below is a detailed breakdown of the most common types:
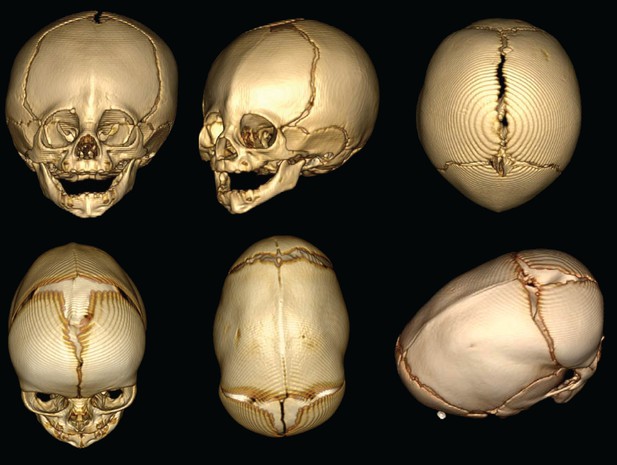
- Sagittal Synostosis: This is the most common type, where the sagittal suture, which runs along the top of the skull from front to back, fuses prematurely. It often results in a long, narrow head shape known as scaphocephaly.
- Coronal Synostosis: In this type, one or both coronal sutures, which run from ear to ear over the top of the head, close too early. If one side is affected, it can lead to an asymmetrical appearance, while bilateral involvement may cause a broad, flat forehead.
- Metopic Synostosis: This involves the premature fusion of the metopic suture, which runs from the nose to the top of the skull. It typically results in a triangular-shaped forehead, known as trigonocephaly.
- Lambdoid Synostosis: This rare form affects the lambdoid suture, located at the back of the skull. It can cause flattening on one side of the back of the head.
Causes of Premature Skull Fusion
The exact cause of craniosynostosis is not always clear, but research has identified several contributing factors. These include genetic mutations, environmental influences, and syndromic associations. Understanding these causes is crucial for both prevention and treatment.
Genetic Factors
In some cases, craniosynostosis is linked to genetic mutations. These mutations may occur spontaneously or be inherited from one or both parents. Syndromic craniosynostosis, where the condition is part of a broader genetic syndrome, is often associated with specific gene abnormalities. Examples include mutations in the FGFR genes, which play a role in bone development.
Environmental Influences
While genetic factors account for many cases, environmental influences can also contribute to the development of craniosynostosis. Maternal health during pregnancy, exposure to certain medications, and nutritional deficiencies have all been studied as potential risk factors. For instance, maternal thyroid disorders or excessive intake of vitamin A during pregnancy may increase the likelihood of premature suture fusion.
Recognizing the Symptoms
Identifying craniosynostosis early is essential for effective management. The symptoms vary depending on the type and severity of the condition but often include visible changes in the shape of the skull. Parents and caregivers should be vigilant for the following signs:
- An abnormally shaped head, such as a long, narrow skull or a flattened area
- A prominent forehead or ridge along the affected suture
- Bulging or sunken fontanelles (soft spots) on the baby’s head
- Delayed closure of the fontanelles
- Developmental delays or neurological issues in severe cases
It is important to note that mild cases may not present noticeable symptoms immediately, making regular pediatric check-ups critical for early detection.
Diagnosing Craniosynostosis
Diagnosing craniosynostosis involves a combination of clinical evaluation, imaging studies, and sometimes genetic testing. Early and accurate diagnosis is key to determining the appropriate treatment plan.
Clinical Evaluation
A healthcare provider will begin by conducting a thorough physical examination of the infant’s head. They will assess the shape of the skull, measure its dimensions, and look for any abnormalities in the sutures or fontanelles. The presence of ridges along the affected sutures or asymmetry in the head shape can be strong indicators of craniosynostosis.
Imaging Studies
To confirm the diagnosis, imaging studies such as X-rays, CT scans, or MRIs may be performed. These tests provide detailed images of the skull and brain, allowing doctors to identify which sutures are fused and assess the extent of the condition. A three-dimensional CT scan is particularly useful for surgical planning, as it offers a comprehensive view of the skull’s anatomy.
Genetic Testing
In cases where craniosynostosis is suspected to be part of a genetic syndrome, genetic testing may be recommended. This involves analyzing the infant’s DNA to identify any mutations or chromosomal abnormalities associated with syndromic craniosynostosis. Genetic counseling may also be offered to families to help them understand the implications and risks of inheritance.
Treatment Options
The treatment for craniosynostosis depends on the type and severity of the condition, as well as the age of the child. In most cases, surgery is required to correct the abnormal skull shape and prevent complications. Non-surgical interventions may also be considered for milder cases or as part of postoperative care.
Surgical Intervention
Surgery is the primary treatment for craniosynostosis and is typically performed within the first year of life. The goal of surgery is to release the fused suture, reshape the skull, and create space for normal brain growth. There are two main surgical approaches:
- Endoscopic Surgery: This minimally invasive procedure is suitable for infants under six months old with single-suture craniosynostosis. Using small incisions and an endoscope, surgeons remove the affected suture and reshape the skull. Postoperative helmet therapy is often required to guide further skull growth.
- Open Cranial Vault Remodeling: This traditional approach is used for more complex cases or older children. It involves making larger incisions to access and reshape the skull bones. While more invasive, it allows for greater precision in correcting severe deformities.
Non-Surgical Approaches
In mild cases or as a complement to surgery, non-surgical treatments may be employed. These include:
- Helmets and Orthotic Devices: Custom-fitted helmets can help mold the skull into a more normal shape after endoscopic surgery or in cases of positional plagiocephaly (a condition often mistaken for craniosynostosis).
- Physical Therapy: Exercises designed to improve neck muscle strength and mobility can help address torticollis, a condition commonly associated with craniosynostosis.
Potential Complications
If left untreated, craniosynostosis can lead to several complications, both physical and neurological. These include:
- Intracranial Pressure: Premature suture fusion can restrict brain growth, leading to increased pressure inside the skull. This can result in headaches, vision problems, and developmental delays.
- Cosmetic Concerns: Abnormal skull shapes can affect a child’s appearance, potentially impacting self-esteem and social interactions as they grow older.
- Neurological Issues: In severe cases, craniosynostosis may interfere with brain development, causing cognitive or motor impairments.
Timely intervention is crucial to minimize these risks and ensure the best possible outcomes for affected children.
Long-Term Outlook
With appropriate treatment, most children with craniosynostosis go on to lead healthy, fulfilling lives. Regular follow-up appointments with a multidisciplinary team, including pediatricians, neurosurgeons, and orthopedic specialists, are essential to monitor progress and address any ongoing concerns. Advances in surgical techniques and postoperative care continue to improve outcomes, offering hope to families facing this challenging condition.