
Wilson’s Disease, often abbreviated as WD, is a rare genetic disorder that affects the body’s ability to regulate copper levels. This condition leads to an excessive buildup of copper in vital organs such as the liver and brain, causing significant health complications if left untreated. In this article, we will explore the intricacies of Wilson’s Disease, including its causes, symptoms, and available treatment options.
Overview of Wilson’s Disease
Wilson’s Disease is a hereditary condition that disrupts the normal processes of copper excretion in the body. Copper is an essential mineral required for various physiological functions, including the production of energy, the formation of red blood cells, and the maintenance of healthy bones and connective tissues. However, when the body accumulates too much copper, it can become toxic and damage critical organs.
The disease was first identified by Dr. Samuel Alexander Kinnier Wilson in 1912, which is why it bears his name. It is estimated to affect approximately one in every thirty thousand individuals worldwide, making it a rare but serious medical condition.
Causes of Wilson’s Disease
Wilson’s Disease is caused by mutations in a specific gene known as ATP7B. This gene is responsible for producing a protein that helps transport copper into bile, a digestive fluid produced by the liver. When the ATP7B gene is defective, the body cannot effectively remove excess copper through bile, leading to its accumulation in the liver and other organs.
Since Wilson’s Disease is inherited in an autosomal recessive pattern, an individual must inherit two copies of the mutated gene—one from each parent—to develop the condition. If a person inherits only one copy of the mutated gene, they are considered a carrier and typically do not exhibit symptoms of the disease.
Risk Factors
- Family History: Having parents or siblings with Wilson’s Disease increases the likelihood of inheriting the condition.
- Ethnicity: Certain populations, such as those of European descent, may have a slightly higher prevalence of the disease.
- Age: Symptoms often appear between the ages of five and thirty-five, although cases outside this range have been reported.
Symptoms of Wilson’s Disease
The symptoms of Wilson’s Disease can vary widely depending on the organs affected and the severity of copper accumulation. Some individuals may experience mild symptoms, while others may face life-threatening complications. The disease can manifest in several ways, including:
Liver-Related Symptoms
- Fatigue and weakness
- Jaundice, characterized by yellowing of the skin and eyes
- Swelling in the abdomen due to fluid accumulation
- Unexplained weight loss
- Tendency to bruise easily
Neurological Symptoms
- Tremors or involuntary shaking of the hands or arms
- Difficulty speaking or slurred speech
- Poor coordination and balance issues
- Muscle stiffness or spasms
- Changes in behavior or personality
Psychiatric Symptoms
- Depression or anxiety
- Impulsive or erratic behavior
- Difficulty concentrating or memory problems
Other Symptoms
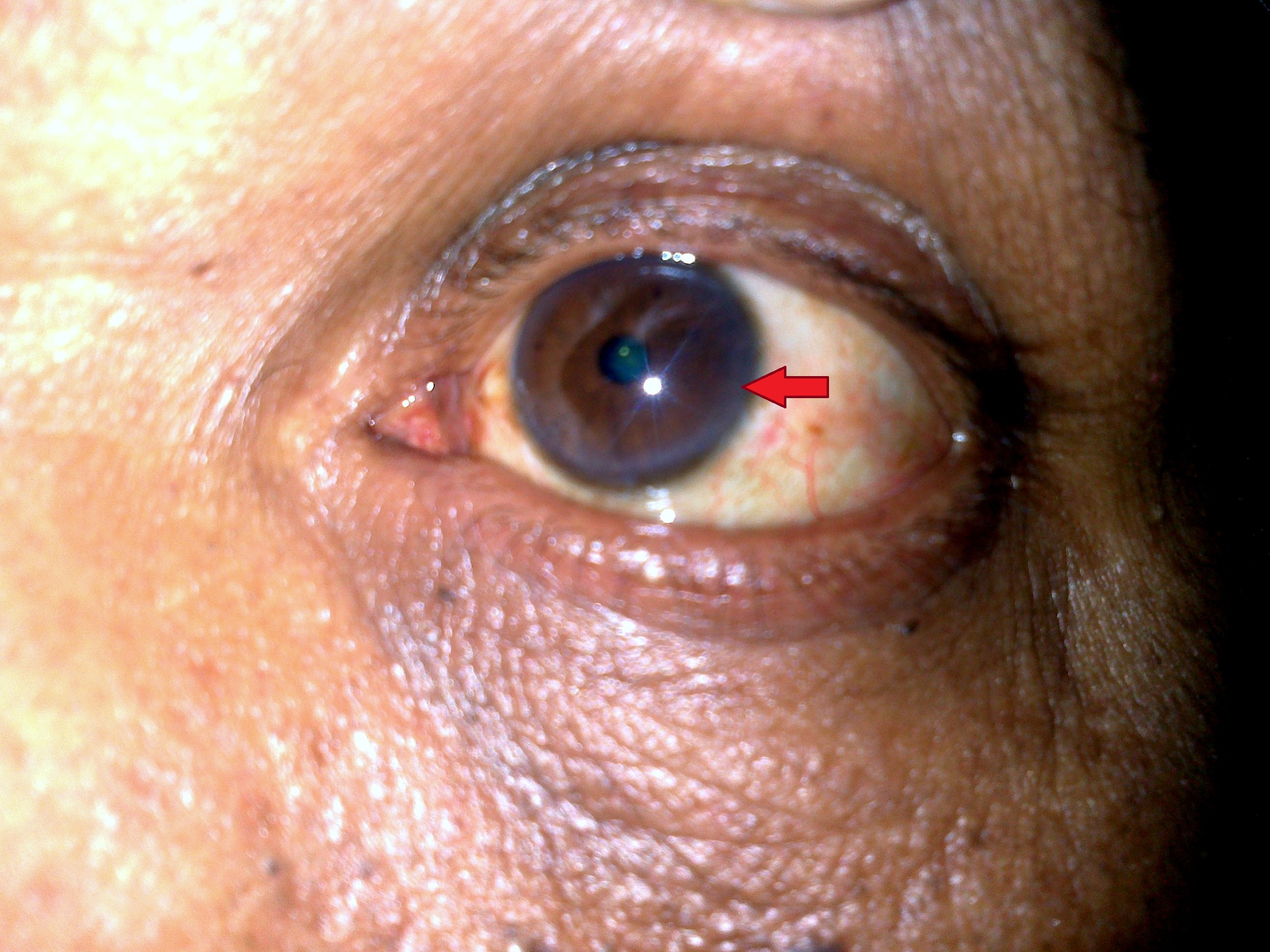
- Kayser-Fleischer rings, which are golden-brown discolorations around the corneas of the eyes caused by copper deposits
- Kidney stones or kidney dysfunction
- Osteoporosis or weakening of the bones
Diagnosis of Wilson’s Disease
Diagnosing Wilson’s Disease can be challenging due to its varied symptoms and rarity. A combination of clinical evaluations, laboratory tests, and imaging studies is often required to confirm the presence of the condition.
Clinical Evaluation
A healthcare provider will begin by reviewing the patient’s medical history and conducting a thorough physical examination. They may look for signs such as Kayser-Fleischer rings or evidence of liver dysfunction.
Laboratory Tests
- Blood Tests: These can measure levels of copper in the blood and assess liver function.
- Urine Tests: A twenty-four-hour urine collection can help determine the amount of copper excreted by the body.
- Liver Biopsy: A small sample of liver tissue may be taken to analyze copper levels and assess liver damage.
Genetic Testing
Genetic testing can identify mutations in the ATP7B gene, providing a definitive diagnosis of Wilson’s Disease. This method is particularly useful for family members of affected individuals who wish to determine their carrier status.
Treatment Options for Wilson’s Disease
While there is no cure for Wilson’s Disease, early diagnosis and treatment can effectively manage the condition and prevent severe complications. The primary goal of treatment is to reduce copper levels in the body and prevent further accumulation.
Medications
Several medications are available to help manage Wilson’s Disease:
- Chelating Agents: These drugs bind to copper and promote its excretion through urine. Examples include penicillamine and trientine.
- Zinc Supplements: Zinc blocks the absorption of copper in the intestines, helping to maintain safe copper levels.
Dietary Modifications
Individuals with Wilson’s Disease are often advised to follow a low-copper diet. Foods high in copper, such as shellfish, nuts, chocolate, and organ meats, should be avoided or consumed in moderation. A registered dietitian can provide personalized guidance to ensure nutritional needs are met while minimizing copper intake.
Liver Transplantation
In severe cases where liver damage is extensive and irreversible, a liver transplant may be necessary. This procedure involves replacing the diseased liver with a healthy donor liver, which can restore normal copper metabolism.
Regular Monitoring
Patients with Wilson’s Disease require lifelong monitoring to ensure that copper levels remain within a safe range. Regular follow-up appointments with a healthcare provider, along with periodic blood and urine tests, are essential components of ongoing care.
Living with Wilson’s Disease
Managing Wilson’s Disease requires a proactive approach to health and wellness. Individuals diagnosed with the condition must adhere to their prescribed treatment plan and make lifestyle adjustments to minimize the risk of complications.
Support Systems
Joining a support group or connecting with others who have Wilson’s Disease can provide emotional support and practical advice. Sharing experiences and coping strategies can help individuals and families navigate the challenges associated with the condition.
Educational Resources
Staying informed about Wilson’s Disease is crucial for effective management. Patients and caregivers should seek out reliable sources of information, such as medical journals, reputable websites, and educational materials provided by healthcare professionals.
Preventive Measures
For individuals with a family history of Wilson’s Disease, genetic counseling can help assess the risk of passing the condition to future generations. Early screening and intervention can significantly improve outcomes for those at risk.