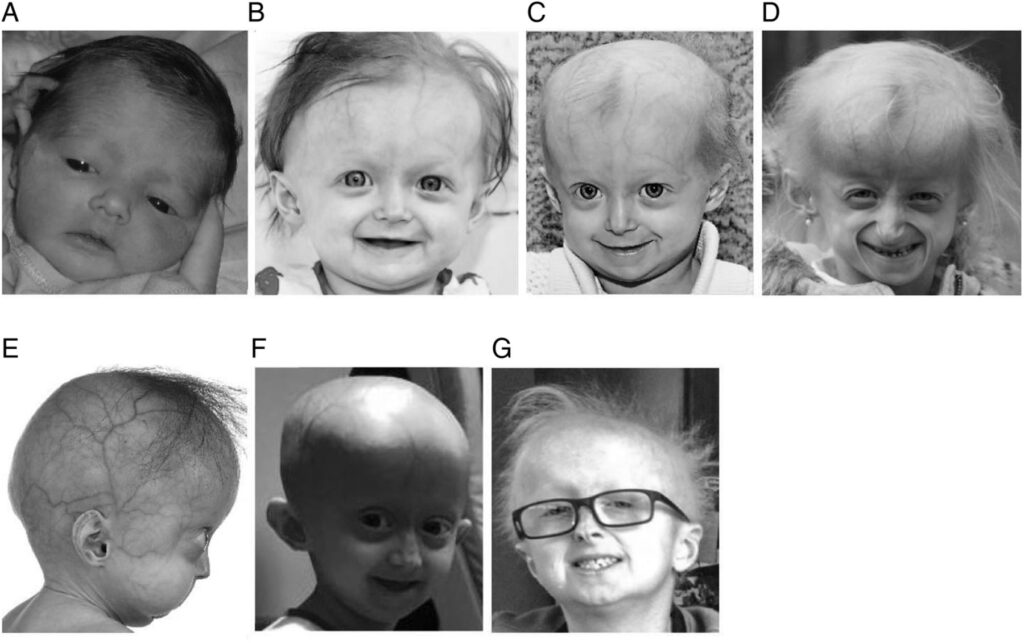

Hutchinson-Gilford Progeria Syndrome, often abbreviated as HGPS, is an extremely rare genetic condition that causes children to age rapidly. This disorder affects approximately one in every four million newborns worldwide. Despite its rarity, the condition has captured significant scientific interest due to its profound impact on affected individuals and their families. Understanding this syndrome requires a closer look at its underlying causes, the symptoms it presents, and the care strategies available for managing its effects.

Understanding the Causes of Hutchinson-Gilford Progeria Syndrome
The primary cause of Hutchinson-Gilford Progeria Syndrome lies in a specific genetic mutation. This mutation occurs in the LMNA gene, which is responsible for producing a protein called lamin A. Lamin A plays a critical role in maintaining the structural integrity of the nucleus within cells. When the LMNA gene is mutated, it produces an abnormal version of lamin A, known as progerin. Progerin accumulates in cells and disrupts their normal function, leading to premature aging.
This genetic mutation is not inherited in the traditional sense. Instead, it arises spontaneously during the early stages of embryo development. As a result, most cases of Hutchinson-Gilford Progeria Syndrome occur sporadically, with no family history of the condition. The exact reasons why this mutation occurs remain unclear, but researchers believe it may be linked to random errors in DNA replication or repair processes.
How the Mutation Affects Cellular Function
- Nuclear Structure: Progerin weakens the nuclear envelope, causing it to become misshapen and unstable. This compromises the cell’s ability to regulate its internal environment.
- Cell Division: Cells with progerin are less efficient at dividing, leading to reduced tissue regeneration and repair.
- Premature Aging: The accumulation of progerin accelerates cellular aging, contributing to the rapid physical decline seen in affected individuals.
Symptoms of Hutchinson-Gilford Progeria Syndrome
The symptoms of Hutchinson-Gilford Progeria Syndrome typically begin to appear within the first two years of life. These symptoms mimic many of the characteristics associated with natural aging, but they occur at an accelerated pace. Early recognition of these signs is crucial for timely diagnosis and intervention.
Physical Characteristics
One of the most noticeable features of Hutchinson-Gilford Progeria Syndrome is the distinct physical appearance it creates. Children with this condition often have:
- A disproportionately small face compared to the size of their head
- Prominent eyes and a thin nose
- A receding chin and thin lips
- Baldness or sparse hair growth
- Visible veins on the scalp due to a lack of subcutaneous fat
Growth and Developmental Issues
In addition to their unique physical traits, children with Hutchinson-Gilford Progeria Syndrome experience stunted growth and developmental delays. Their height and weight often fall below average for their age group. Joint stiffness and limited mobility are also common, making it difficult for them to engage in physical activities typical for their peers.
Health Complications
As the condition progresses, affected individuals face a range of serious health complications. These include:
- Cardiovascular problems such as hardening of the arteries, which can lead to heart attacks or strokes
- Osteoporosis, resulting in fragile bones prone to fractures
- Hip dislocations and other musculoskeletal issues
- Hearing loss and vision impairments
Caring for Individuals with Hutchinson-Gilford Progeria Syndrome
While there is currently no cure for Hutchinson-Gilford Progeria Syndrome, various treatment approaches aim to manage symptoms and improve quality of life. Care for affected individuals requires a multidisciplinary approach involving medical professionals, therapists, and caregivers.
Medical Interventions
Medical treatments focus on addressing the specific health challenges posed by the condition. Some of the key interventions include:
- Cardiovascular Care: Regular monitoring and management of heart health are essential. Medications such as statins and anticoagulants may be prescribed to reduce the risk of heart disease.
- Hormone Therapy: Growth hormone treatments can help improve height and weight gain in some cases.
- Bone Health: Supplements like calcium and vitamin D, along with medications to strengthen bones, are often recommended to combat osteoporosis.
Physical and Occupational Therapy
Physical therapy plays a vital role in maintaining mobility and preventing joint stiffness. Therapists work with patients to develop exercises tailored to their abilities, helping them stay active and independent for as long as possible. Occupational therapy focuses on enhancing daily living skills, ensuring that individuals can perform tasks such as dressing, eating, and grooming with minimal assistance.
Psychological and Emotional Support
Living with Hutchinson-Gilford Progeria Syndrome can be emotionally challenging for both patients and their families. Counseling services provide a safe space to address feelings of anxiety, depression, or isolation. Support groups connect families facing similar challenges, fostering a sense of community and shared understanding.
Nutritional Guidance
A balanced diet is crucial for supporting overall health and well-being. Nutritionists collaborate with families to create meal plans that meet the unique dietary needs of individuals with this condition. Adequate caloric intake, hydration, and nutrient-rich foods are emphasized to promote optimal growth and energy levels.
Tips for Nutritional Management
- Include high-calorie snacks to address weight-related concerns
- Focus on foods rich in vitamins and minerals to support bone and immune health
- Encourage frequent, small meals if appetite is limited
Advancements in Research and Future Prospects
Although Hutchinson-Gilford Progeria Syndrome remains incurable, ongoing research offers hope for improved treatments and potential breakthroughs. Scientists are exploring innovative therapies aimed at targeting the root cause of the condition—the abnormal production of progerin.
Experimental Treatments
Recent studies have investigated the use of farnesyltransferase inhibitors, a class of drugs originally developed for cancer treatment. These medications block the attachment of progerin to the nuclear membrane, potentially slowing down its harmful effects. Clinical trials have shown promising results, with some participants experiencing modest improvements in weight gain and cardiovascular health.
Gene Editing Technologies
Emerging technologies such as CRISPR-Cas9 hold immense promise for correcting the genetic mutation responsible for Hutchinson-Gilford Progeria Syndrome. By precisely editing the LMNA gene, scientists hope to prevent the production of progerin altogether. While still in experimental stages, these advancements could pave the way for groundbreaking treatments in the future.
Raising Awareness and Funding
Increased awareness of Hutchinson-Gilford Progeria Syndrome has led to greater funding opportunities for research initiatives. Nonprofit organizations and advocacy groups play a pivotal role in educating the public, organizing fundraising events, and supporting affected families. Continued efforts in this area are essential for driving progress and improving outcomes for individuals with this condition.